Development of methods to prepare various organic compounds of synthetic, biological, and medicinal significance in a cost-effective, efficient, reliable and selective manner is vital to advances in human health care.
The central theme of my research program is the development of synthetic methods that possess these desirable attributes and allow rapid access to the important molecules from an array of readily accessible but otherwise chemically inert starting materials.
Synthesis of a target molecule can often be accelerated through the use of a catalyst system; the catalysts facilitate the union of poorly reactive starting materials by converting them into a set of highly reactive intermediates en route to the products.
Nonetheless, key shortcomings limit the application of catalytic methods in complex molecule synthesis.
Among the critical unresolved issues in the state-of-the-art is the catalyst deactivation, occurring through the formation of stable adducts within a reaction mixture of the catalysts, starting materials, intermediates and/or products.
Such process can either result in deceleration or termination of the target reaction.
To suppress catalyst deactivation, the range of starting materials is confined to those that pose minimum risk of generating the inert adducts.
The aim of my research team is to address these problems through the design and development of highly efficient and multi-functional catalyst systems that cannot be easily disabled/quenched.
We aspire to demonstrate that our catalysts may be used for direct conversion of complex natural products, pharmaceuticals, and agrochemicals to a variety of derivatives of interest.
Along these lines, we have achieved the following during the past five years:
(1) We have introduced potent and versatile catalyst systems that can activate an assortment of amines, ethers, carbonyl-containing and peptide-based compounds, thereby allowing rapid access to their useful derivatives.
(2) We have developed pathways that convert products generated by the catalytic reactions to different valuable products.
(3) We have carried out detailed mechanistic studies to elucidate how the catalysts operate, hoping to use the resulting knowledge to further our work in the future.
Cooperative Catalysts for Enantioselective Transformations of N-Alkylamines
A desirable organic molecule can be synthesized more efficiently through the use of various combinations of chiral Lewis acid and/or Lewis base catalysts that are designed to function cooperatively (i.e., cooperative catalysts). 1-3A key advantage of a multi-catalytic process is that it facilitates an otherwise-difficult-to-perform union of non isolable intermediates (e.g., 1a → iminium I or enamine II → 2a or 3a; Figure 1) under neutral pH conditions. 4-5 The wasteful pre-activation step may thus be obviated. Nevertheless, notable shortcomings remain unaddressed. 1-3 For instance, self-quenching might occur in a mixture that contains an electrophile, a nucleophile, together with an acid and a base catalyst (e.g., 1a → classic Lewis adduct). One way to circumvent acid–base complexation is by avoiding a combination that exhibits high affinity (i.e., hard–hard or soft–soft pairing). However, this latter approach has thus far been limited to cases in which weakly to moderately acidic and/or basic promoters are involved, where only highly acid- or base-sensitive substrates can be used. Development of potent and unquenchable cooperative multi-catalyst systems that facilitate reactions between unactivated substrates is an important and largely unresolved problem in enantioselective catalysis.
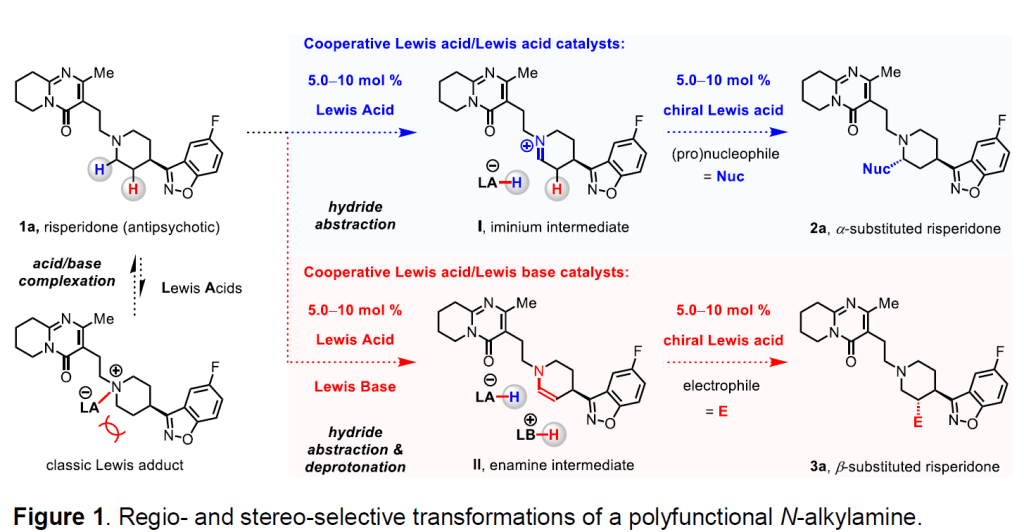
The application of frustrated Lewis pairs (FLPs), consisting of hindered and electronically disparate Lewis acids and Lewis bases, has emerged as an attractive strategy for overcoming mutual quenching. 6 An unquenched acid/base pair may be utilized for synergistic activation of otherwise unreactive small molecules, such as H 2 and CO 2 . 8, 9 Additionally, FLPs that are comprised of a boron-based Lewis acid and a Lewis basic amine substrate bearing easily accessible α-hydrogens, have been shown to engage in Lewis acid-mediated hydride abstraction; such processes produce an iminium cation and a borohydride anion (e.g., 1a → I, Figure 1). 10, 11 When we began our investigations (2016), engagement of iminium ions formed by organoborane-catalyzed hydride abstraction from amines in the context of a catalytic reaction was largely limited to dehydrogenation of N-containing heterocycles, 12 and these strategies were yet to be successfully applied to catalytic and enantioselective C–C bond forming processes. One way to address this problem would be to design a catalyst system which can abstract a hydride from an amine substrate, mediate enantioselective C–C bond formation, and then regenerate the active Lewis pair without significant deactivation mediated by a Lewis basic moiety.
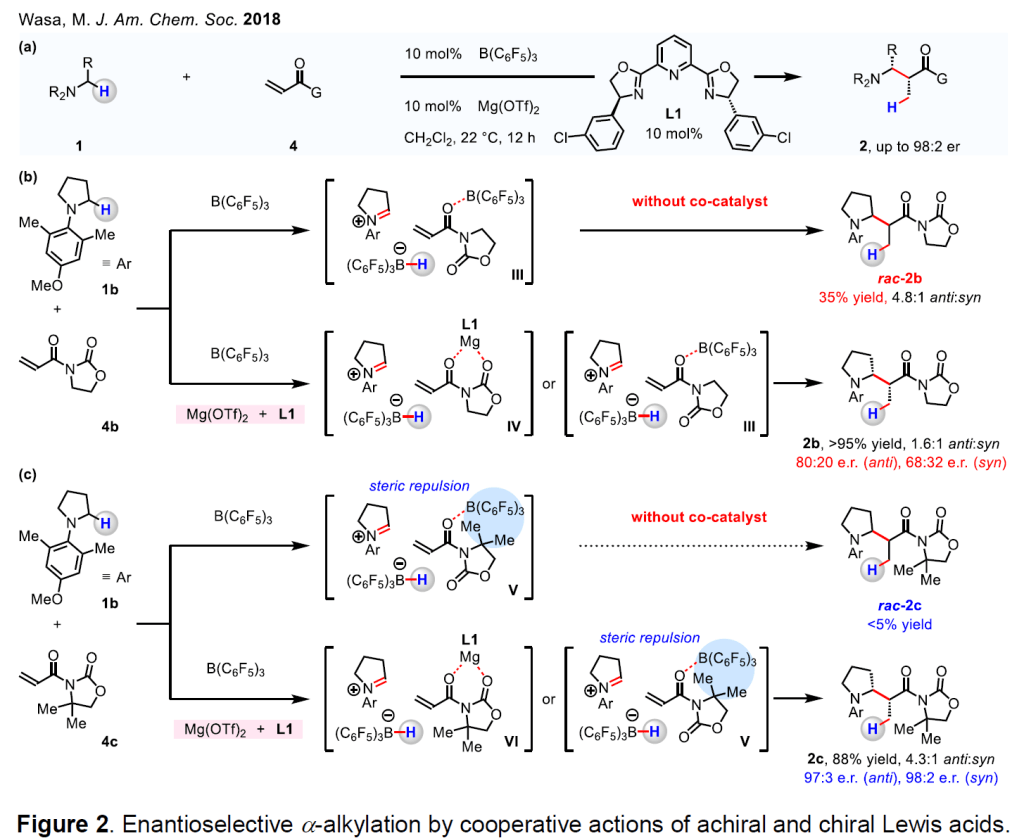
We have developed a method for the enantioselective union of various N-alkylamines (1) and α, β-unsaturated compounds (4) by implementing the cooperative action of two untethered and independently operational Lewis acid catalysts (Figure 2). 13 One key advantage of the approach would be that efficiency and stereoselectivity might be optimized through evaluation of pairs of readily accessible Lewis acids and chiral ligands (vs bifunctional catalysts needing cumbersome tethering of the catalyst units). 1-3 A central design principle is that the untethered Lewis acid catalysts must perform their separate tasks without any overlapping of their function, as otherwise, stereoselectivity would suffer (owing to reaction facilitated by the achiral component). 1-3 Specifically, we illustrated that by properly tuning the structural and electronic features of different Lewis acids and substrates, the ability of B(C 6 F 5 ) 3 and chiral (TfO) 2 Mg/L1 to serve as a hydride acceptor from 1, or an activator of 4, can be adjusted. We found out that the racemic C–C bond forming reaction of 1b and 4b can be catalyzed by B(C 6 F 5 ) 3 alone (1b and 4b → III → rac–2b, Figure 2b). As a result of this competing racemic process, 2b can be obtained with only up to 80:20 er when the union of 1b and 4b is catalyzed by B(C 6 F 5 ) 3 and (TfO) 2 Mg/L1(2b is formed via either III → rac–2b or IV → 2b). To improve enantioselectivity further, we reacted1b with a more sterically hindered electrophile 4c (vs 4b), which resulted in the formation of 2c in up to 98:2 er (Figure 2c). The improved enantioselectivity likely arises from a preference for Mg-activated VI over (F 5 C 6 ) 3 B-activated V, the latter of which suffers from severe steric pressure between the gem-dimethyl group of 4c and B(C 6 F 5 ) 3 . This contention is supported by the fact that 1b and 4c do not react in the absence of (TfO) 2 Mg/L1. The principles delineated by our work later served as the conceptual framework for the development of new processes that demand separate and independently operational Lewis acidic co-catalysts whose functions overlap and the use of which initially seems to have a negative impact on enantioselectivity.
In subsequent work, we outlined an approach for activation of α-amino C–H bonds to generate propargyl amines (Figure 3). 14 We discovered that by using a blend of B(C 6 F 5 ) 3 and an organocopper complex, it is possible to generate an iminium from an N-alkylamine and a L n Cu–alkynyl complex from an alkynylsilane (1 and 5→ VII). This catalyst system tolerates a wide variety of Lewis acid-sensitive functional groups, and is therefore applicable to the late- stage transformations of complex (and bioactive) trialkyl amines to propargylamines (e.g., 2d–2g; Figure 3a). Furthermore, by using B(C 6 F 5 ) 3 and a chiral L n Cu–PyBOX complex, we established that highly enantioselective synthesis of propargylamines can be achieved (e.g., 1h → 2h, Figure3b). Mechanistic investigations involving the determination of rate orders and kinetic isotope effects were performed to elucidate how the two co-catalysts operate cooperatively.

Transformations of β-Amino C–H Bonds by Cooperative Acid/Base Catalysis
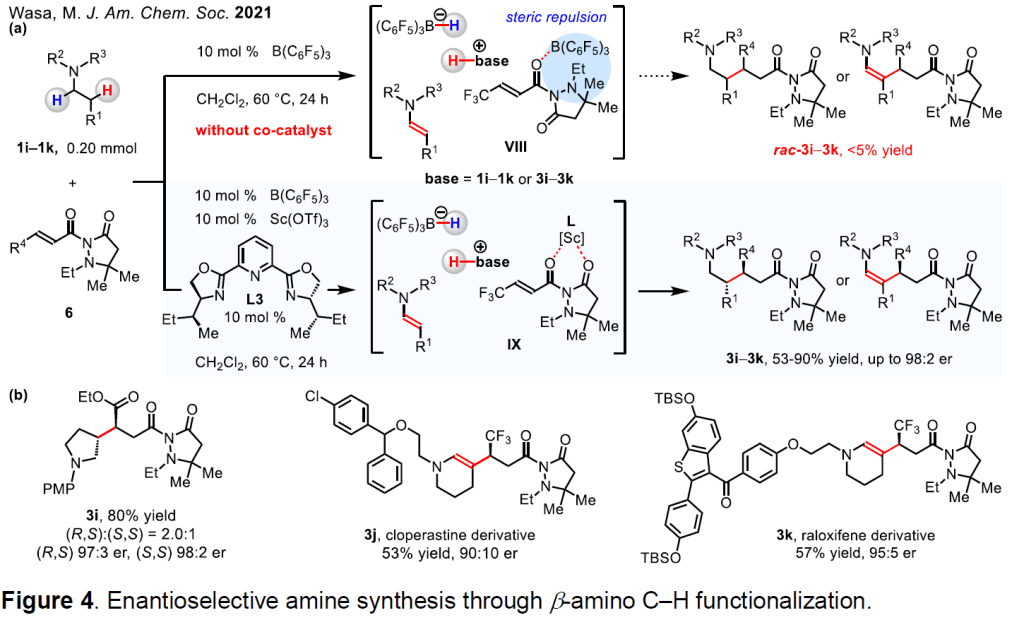
The development of methods for enantioselective synthesis amines by activation of the difficult-to-access β-amino C–H bond is a major and unresolved challenge. 15 α-Amino C–H activation by hydrogen atom or hydride transfer is facilitated by the stabilization of the resulting species through electron delocalization from the nitrogen lone pair (e.g., amine → iminium ion). 15 Still, such processes do not readily occur at the β position of amines. We have discovered that a potent catalyst system comprised of B(C 6 F 5 ) 3 , a chiral Sc-based co-catalyst, and a Brønsted base promotes enantio- and diastereo-selective reactions of N-alkylamines (e.g., 1i–1k) and α, β-unsaturated compounds (6) to give β-alkylated amines (3i–3k). 16 Our mechanistic studies have unveiled that B(C 6 F 5 ) 3 and the Brønsted base (e.g., amines 1 and/or 3) convert 1 into an enamine via sequential hydride abstraction and deprotonation (1 → VIII or 1 → IX). We have found that B(C 6 F 5 ) 3 alone is incapable of promoting the union of 1 and 6 to afford racemic products (VIII → rac-3i–3k). However, in the presence of the Sc-based complex, C–C bond formation occurs smoothly (IX → 3i–3k). A variety of Lewis acid-sensitive functional units are tolerated, and, as a result, the approach is applicable to late-stage modification of relatively complex (and bioactive) trialkylamines (e.g., 3j and 3k).
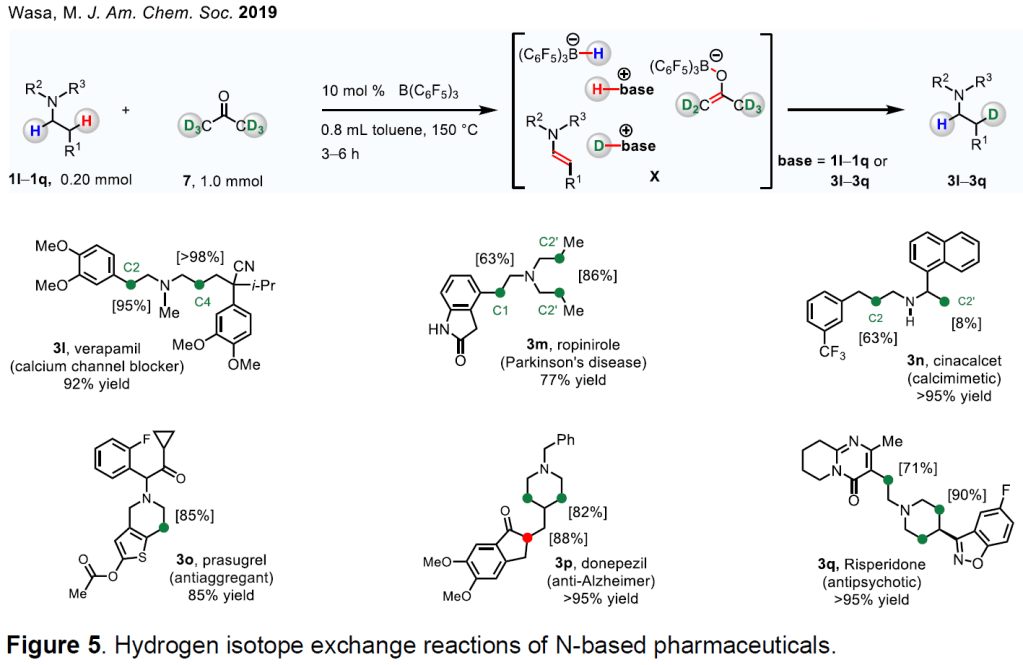
We have designed a method for site-selective deuterium labeling of β-amino C–H bonds in bioactive amines (Figure 5). 17 By implementing the cooperative action of B(C 6 F 5 ) 3 and amine, we have illustrated that the N-alkylamine-based drugs containing different functional groups can be converted into the corresponding enamine (1l–1q → X), and that the same catalyst system can generate a labeling agent from acetone-d 6 by removal of deuterium (7 → X). The ensuing reaction of the enamine and deuterating reagent affords the drug isotopologues (e.g., 3l–3q).
Transformations of Polyfunctional Ethers and Peptides by Cooperative Catalysis
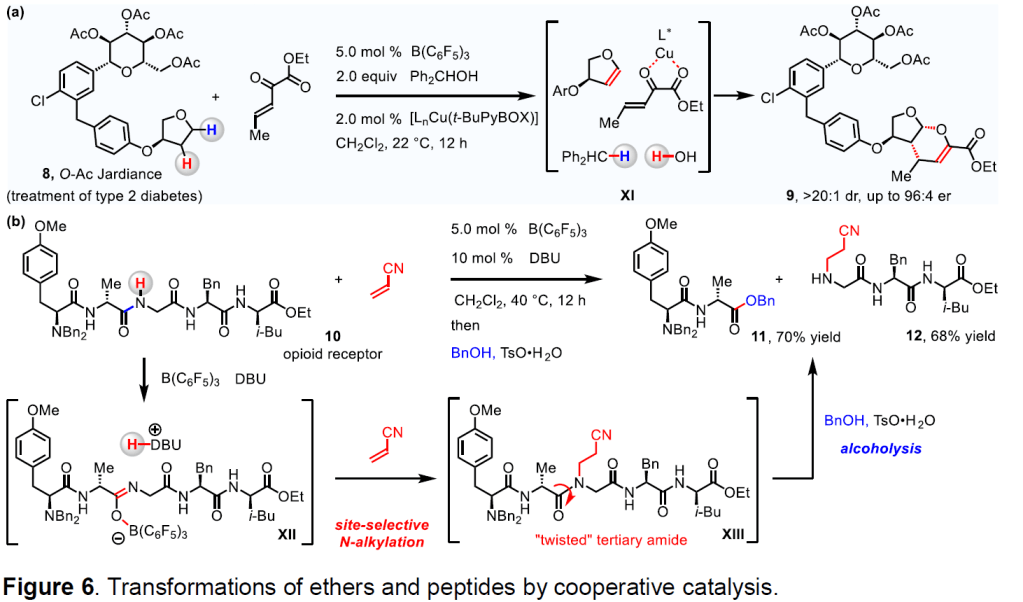
We are engaged in the development of enantioselective reactions of vinyl ethers formed by C–H activation of an ether moiety (Figure 6a). A cation, formed by the reaction of B(C 6 F 5 ) 3 and an alcohol, abstracts a hydride from the ether substrates to afford oxocarbenium ions that may be deprotonated to furnish vinyl ethers (e.g., 8 → XI). A chiral Cu-based co-catalyst promotes the stereoselective inverse electron demand Diels-Alder reaction between ethyl 2-oxopent-3-enoate and the vinyl ether (XI → 9). The stereoselective merger of ethers and various coupling partners is expected to be highly applicable to complex carbohydrate and natural product syntheses.
Strategies for site-selective C–N bond cleavage of peptides, proteins, and other amide- based compounds are crucial to our ability in determining their respective structures, to studies in biological chemistry, as well as to drug development. 18 Accordingly, we are investigating the regioselective conversions of C–N bonds within polyfunctional peptides into C–C, C–N and C–O bonds (Figure 6b). A representative process entails the use of B(C 6 F 5 ) 3 and DBU to deprotonate the most sterically accessible glycine residue of a pentapeptide to form an amidate (10 → XII); the following N-alkylation by acrylonitrile yields a tertiary amide (XII → XIII). Because the C–N bond of the sterically hindered and thereby “twisted” tertiary amide unit is weaker than those of the 3 secondary amides, XIII undergoes site-selective alcoholysis to afford an ester and an amine (XIII → 12 and 13).
In summary, we have developed cooperative catalyst systems that enable structural modification and diversification of an array of polyfunctional molecules at positions that are difficult to access by previously established methods. We have demonstrated that these highly reactive and versatile catalysts can activate C–H or N–H bonds of complex amines, ethers or peptides and subsequently promote transformations of the resulting intermediates. Further catalyst development is likely to lead to additional discoveries regarding generally applicable strategies for natural product synthesis, drug discovery, and determination of the identity of complex molecules.
References (please see the attached PDF)

The Herbert Wertheim UF Scripps Institute for Biomedical Innovation & Technology
130 Scripps Way, Jupiter, FL 33458
Email: wasam@ufl.edu
